Research
Mitochondrial Trafficking and Quality Control

Movie: Mitochondria trafficking in a rat cortical neuron
Due to their unique morphology, neurons have a special need to transport organelles such as mitochondria throughout their arbors, wherever their functions are required. Mitochondria travel long distances to meet the energy supply and calcium buffering demands even in the most distal parts of the neuron. Failure to transport mitochondria has been associated with many neurodegenerative disorders, so understanding the proteins involved in the transport and the signaling pathways regulating them is of foremost importance. Our lab focuses on the outer mitochondrial Rho GTPases (MIRO1/2) and the trafficking kinesin (TRAK1/2). These molecular adaptors serve as hubs for the intracellular signaling that modulates mitochondrial motility. We are currently studying the structure of this complex, how MIRO1 GTPase domains regulate the assembly of the complex, and how the TRAK1 adaptor induces mitochondrial arrest upon AMPK signaling. By understanding those processes, we gain insight into how trafficking defects contribute to neurological disorders.
Axonal RNA Transport
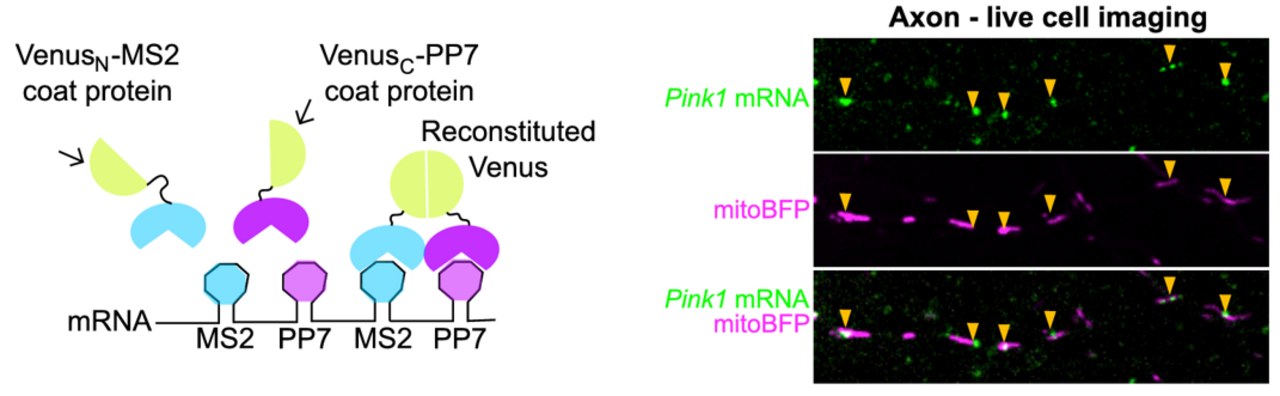
The Schwarz lab has expanded its focus on mitochondrial function in neurons to include mRNAs co-trafficked on the outer mitochondrial membrane (OMM). We began with a candidate approach, studying how PINK1 mRNA is transported to support local mitophagy (Harbauer et al. 2022). The PINK1 protein, a kinase crucial to initiating a signal cascade that results in autophagosome engulfment of dysfunctional mitochondria, has an extremely short half-life. Since this seconds- to minutes-long protein half-life is incompatible with transport into distal dendrites and axons, we reasoned the mRNA must be locally transported. We used live mRNA imaging to show that PINK1 mRNA is indeed colocalized with mitochondrial in distal neurites. We identified Synaptojanin2 as a key protein localizaing mRNA to the OMM. Synaptojanin2 achieves this via an RNA Recognition Motif (RRM) that binds mRNA, and a PDZ domain that interacts with the OMM protein SYNJ2BP. Knockdown of Synaptojanin2 or SYNJ2BP disrupts local depolarization-induced mitophagy in primary rodent neurons, likely by preventing local PINK1 mRNA trafficking.
Synaptojanin2 appears to bind many other species of mRNA and we are using mice and iPSC cell lines with mutations in the RRM to further dissect the role of Synaptojanin2 in neuronal mRNA trafficking. Inspired by recent proximity ligation techniques (e.g. Fazal et al. 2019), we have begun cataloging mRNAs enriched on the neuronal OMM via RNAseq. We are also interested in nucleotide motifs that may mediate anchoring to mitochondria.
Function of Kinetochore in Neurons
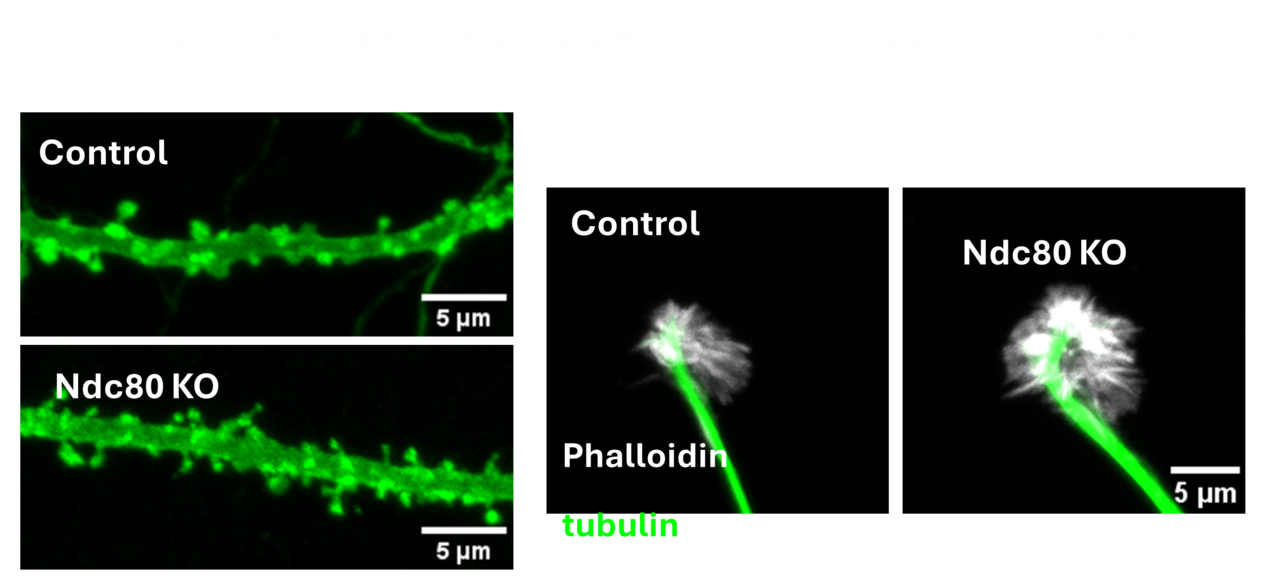
The kinetochore is a protein complex that has been studied intensively for its crucial role in chromosome mechanics during cell division. Surprisingly the kinetochore complex also has an essential function in postmitotic neurons – a population that famously never undergoes cell division. Neurons, however, like dividing cells require careful regulation of the microtubule cytoskeleton and neurons appear to have repurposed the kinetochore complex for controlling microtubule dynamics. We seek to understand the role of kinetochore proteins in the formation of synaptic terminals, dendrites, dendritic spines and axon growth. This project leverages the opportunities of both conditional knock out mice and mammalian primary cultures to elucidate the mechanism by which these proteins function in neurodevelopment and the sculpting of neuronal connections.
Mitochondrial Dysfunctions and Neurodegeneration
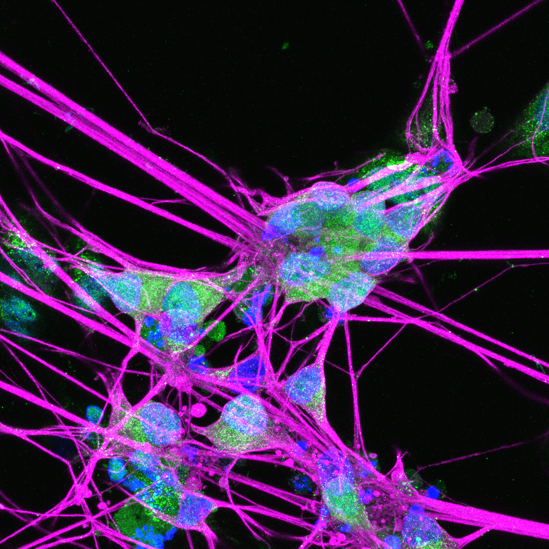 Mitochondrial dysfunctions cause neurological diseases. Our lab studies Autosomal Dominant Optic Atrophy (ADOA), the most prevalent form of hereditary optic neuropathy, which is caused by mutations in the OPA1 gene. The OPA1 protein regulates key mitochondrial functions including inner membrane fusion and cristae maintenance. Heterozygous mutations in OPA1 lead to selective death of retinal ganglion cells (RGC) and vision loss in ADOA patients. We have built a novel mouse model carrying a pathogenic Opa1 mutation that recapitulates key features of ADOA. We discovered that loss of the neurodegeneration trigger, SARM1, prevents RGC death and vision decline in the Opa1 mutant mice. We are currently developing SARM1-based therapies towards ADOA and other mitochondrial neuropathies.
Mitochondrial dysfunctions cause neurological diseases. Our lab studies Autosomal Dominant Optic Atrophy (ADOA), the most prevalent form of hereditary optic neuropathy, which is caused by mutations in the OPA1 gene. The OPA1 protein regulates key mitochondrial functions including inner membrane fusion and cristae maintenance. Heterozygous mutations in OPA1 lead to selective death of retinal ganglion cells (RGC) and vision loss in ADOA patients. We have built a novel mouse model carrying a pathogenic Opa1 mutation that recapitulates key features of ADOA. We discovered that loss of the neurodegeneration trigger, SARM1, prevents RGC death and vision decline in the Opa1 mutant mice. We are currently developing SARM1-based therapies towards ADOA and other mitochondrial neuropathies.
Picture: human RGCs differentiated from OPA1 mutant iPSCs